パラメータ不要の表現が単一細胞基盤モデルを下流ベンチマークで上回るという主張と、その根拠
単一細胞RNAシーケンスの代表的な下流ベンチマークでは、大規模な基盤モデルの埋め込みを使わなくても、細胞内正規化と線形手法を中心にした単純で解釈可能な表現で最先端級、またはそれに近い性能に到達できると示しています。
TL;DR(結論)
- 単一細胞RNAシーケンスの代表的な下流ベンチマークでは、大規模な基盤モデルの埋め込みを使わなくても、細胞内正規化と線形手法を中心にした単純で解釈可能な表現で最先端級、またはそれに近い性能に到達できると示しています。
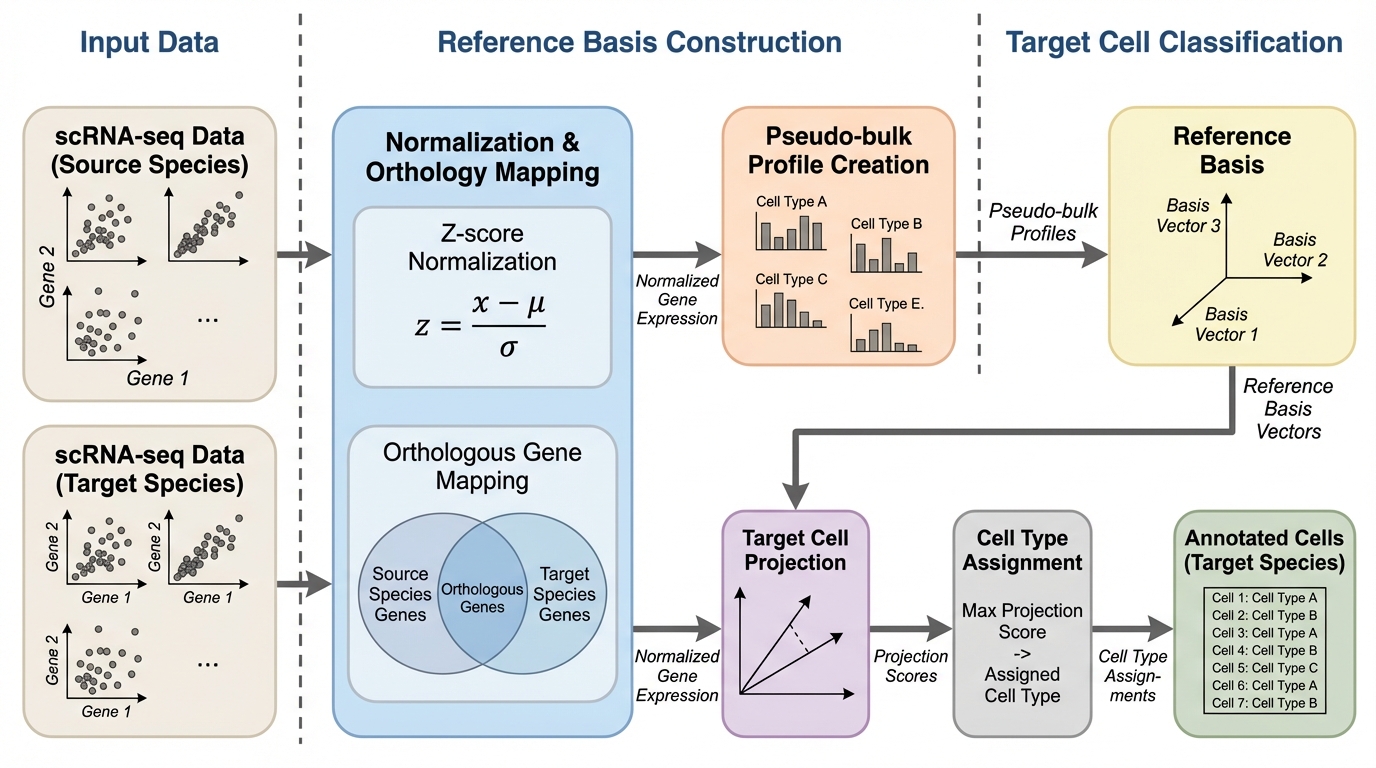
- 遺伝子発現を「細胞ごとに独立に」順位にもとづくスコアへ正規化し、scTOPによる線形射影、あるいはANOVAによる遺伝子選択とPCAでの次元削減を経てロジスティック回帰へ入れる手順で、種間アノテーションやヒト組織データの細胞型分類などを比較しています。
- 分布外の設定(学習データにない細胞型や生物種を含む条件)でも基盤モデルを上回る場面があるため、現行ベンチマークは複雑な非線形表現の「発見」より、正規化済み遺伝子発現ベクトルが持つ低複雑な構造を測っている可能性があると結論づけています。
なぜこの問題か
単一細胞RNAシーケンス(scRNA-seq)には、強く再現性のある統計的構造があるとされてきました。その期待が、大規模データ(細胞アトラスのような集積)を背景に、TranscriptFormerのようなトランスフォーマー系アーキテクチャで遺伝子を潜在空間へ埋め込み、遺伝子発現(mRNAカウント)の生成モデルを学習する「基盤モデル」を後押ししてきました。これらの埋め込みは、細胞型分類、疾患状態予測、種をまたぐ学習などの下流タスクで最先端(SOTA)級の性能が報告され、汎用的な生物学的表現を学んだ証拠として引用されることもあります。 一方で、基盤モデルが得ている表現が、適切に処理したscRNA-seqデータに元々含まれている構造を超えているのかどうかは明確ではないと述べられています。実際、scTOPのような線形で解釈可能な方法が、多様な単一細胞タスクで驚くほど良い性能を示すという流れも紹介されています。著者らはこの状況を踏まえ、「scRNA-seqデータ自体の構造はどれほど複雑なのか」「現在よく使われるベンチマークが要求する表現の高度さはどの程度なのか」という基本的な問いを改めて立てています。…
核心:何を提案したのか
本論文の中心的な提案は、計算コストが高い深層学習ベースの表現に依存せずとも、注意深い前処理と正規化、そして線形手法にもとづく単純で解釈可能なパイプラインによって、単一細胞基盤モデルがよく評価される複数のベンチマークで最先端級、またはそれに近い性能を得られるという点です。ここでの立場は、細胞を「遺伝子発現空間のベクトル」として扱い、線形代数や線形分類器で下流タスクを解くという整理になっています。 具体的には、パラメータ不要の線形代数的手法としてscTOP(Single-cell Type Order Parameters)が示され、種間の細胞型アノテーションにおいて基盤モデル(TF-Exemplar、TFMetazoaとして報告されている結果)より高いmacro F…
続きはログイン/プランで閲覧できます。
続きを読む
ログインで全文を月 2 本まで無料で読めます
無料プランで全文は月 2 本まで読めます。
Related